
Zespół Angelmana jest złożonym genetycznie defektem, który przede wszystkim afektuje system nerwowy.
Charakterystyczne objawy to: opóźnienie w rozwoju, intelektualne inwalidztwo, upośledzenie mowy, problemy w poruszaniu sie i zaburzenia równowagi.
Większość chorych na te chorobę dzieci posiada regularne napady drgawek (epilepsja) oraz małogłowie (microcephaly).
Opóźniony rozwój staje sie zauważalny miedzy 6-tym, a 12-tym miesiącem życia.
Inne sygnały i symptomy ujawniają sie stopniowo we wczesnym dzieciństwie.
Dzieci z Zespołem Angelmana są zazwyczaj radosne, nadpobudliwe i często bezzasadnie wybuchają śmiechem.
Charakteryzuje je nadpobudliwość, krotka koncentracja i trzepotanie rękoma.
Większość chorych dzieci ma również problemy ze snem i potrzebuje mniej snu niż ich zdrowi rówieśnicy.
Niektóre z nich są niezwykle jasnej karnacji i maja zdecydowanie jasne włosy
Wraz z wiekiem osoby z zespołem Angelmana staja sie mniej pobudliwe, i maleją problemy ze snem.
Osoby dotknięte ta choroba pozostają upośledzone umysłowo i fizycznie przez całe życie.
Maja ciężkie zaburzenia mowy oraz napady epileptyczne.
Jak rozpowszechniony jest Zespół Angelmana?
Zespół Angelmana dotyka od 1 na 12,000 do 1 na 20,000 dzieci.
Jakie genetyczne defekty wywołuje zespół Angelmana?
Wiele charakterystycznych cech Zespołu Angelmana wynika z utraty funkcji genu UBE3A.
Ludzie zwykle dziedziczą jedna kopie genu UBE3A od każdego z rodziców.
Obie kopie tego genu są aktywne w wielu tkankach organizmu.
W pewnych obszarach mózgu, jednak tylko pochodząca od matki kopia genu UBE3A jest aktywna.
Jeśli matczyna kopia genu UBE3A została utracona lub jest niezdolna do prawidłowego funkcjonowania np. z powodu mutacji, wówczas nie będzie żadnej aktywnej kopii genu w określonych partiach mózgu.
-------------------------------------------
Istnieje kilka rożnych przyczyn dezaktywacji matczynej kopii genu UBE3A.
Większość przypadków zespołu Angelmana (około 70 %) występuje, gdy segment chromosomu 15 zawierający gen UBE3A został utracony.
W innych przypadkach (ok. 11 %), zespół Angelmana spowodowany jest mutacja w matczynej kopi tego genu.
W niewielkich ilości przypadków, osoby z zespołem Angelmana odziedziczyły obie kopie chromosomu 15 od ojca, •zamiast po jednym egzemplarzu od każdego rodzica. Zjawisko to nazywa sie ojcowska disomia jednorodzicielska.
Rzadko, zespół Angelmana może być również spowodowany przez przegrupowanie chromosomów (relokacje), •lub mutacje lub inny defekt w rejonie DNA, który odpowiada za aktywacje genu UBE3A.
Dotyczy to zmian genetycznych, które mogą wyłączyć (zdezaktywować) matczyny gen UBE3A.
W 10% - 15 % przypadków przyczyny choroby nie da sie określić. W tych przypadkach zmiany dotyczące innych genów i chromosomów mogą być odpowiedzialne za wystąpienie tej choroby.
-------------------------------------------
Gen OCA2 znajduje sie na odcinku chromosomu 15, który jest często nieaktywny u osób z Zespołem Angelmana.
Białko produkowane na podstawie tego genu pozwala dokończyć pigmentacje skory, włosów i oczu.
Z utrata genu zwanego OCA2 związane są: jasna karnacja skory i jasne włosy.
Więcej informacji na temat : OCA2, genu UBE3A i chromosomu 15.
Czy Zespól Angelmana jest choroba dziedziczna?
Większość przypadków Zespołu Angelmana nie jest dziedziczne, a w szczególności te, •które wynikają z wypadnięcia genu UBE3A w matczynym chromosomie 15 lub disomia ojcowska chromosomu 15.
Te genetyczne zmiany maja charakter przypadkowy.
Choroba dotyka ludzi, których rodzinna historia nie notuje żadnych podobnych przypadków.
-------------------------------------------
Rzadko zmiany genetyczne odpowiedzialne za zespół Angelmana mogą być dziedziczone.
Na przykład możliwe jest dziedziczenie mutacji w genie UBE3A lub dziedziczenie zmian w DNA
dezaktywujących matczyny gen UBE3A.
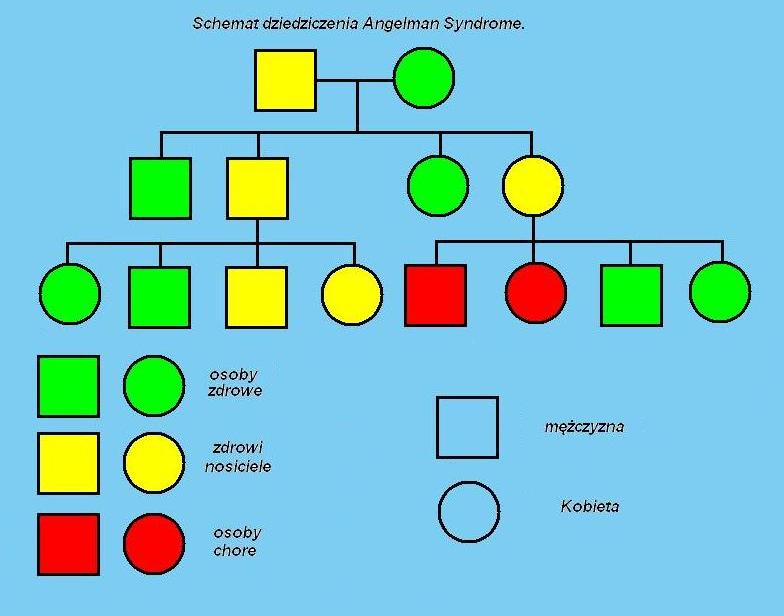
Schemat dziedziczenia Zespołu Angelmana. (2003 Oulu University Library)
Jak przetłumaczyć napisy na filmie; wystartuj film, włącz napisy (zobacz poniżej), wybierz język.

Brak komentarzy:
Prześlij komentarz